No Evidence of Disease Activity (NEDA-3) in the Phase 3 EVOLVE-MS-1 Study
NEDA-3 Study Graphic*
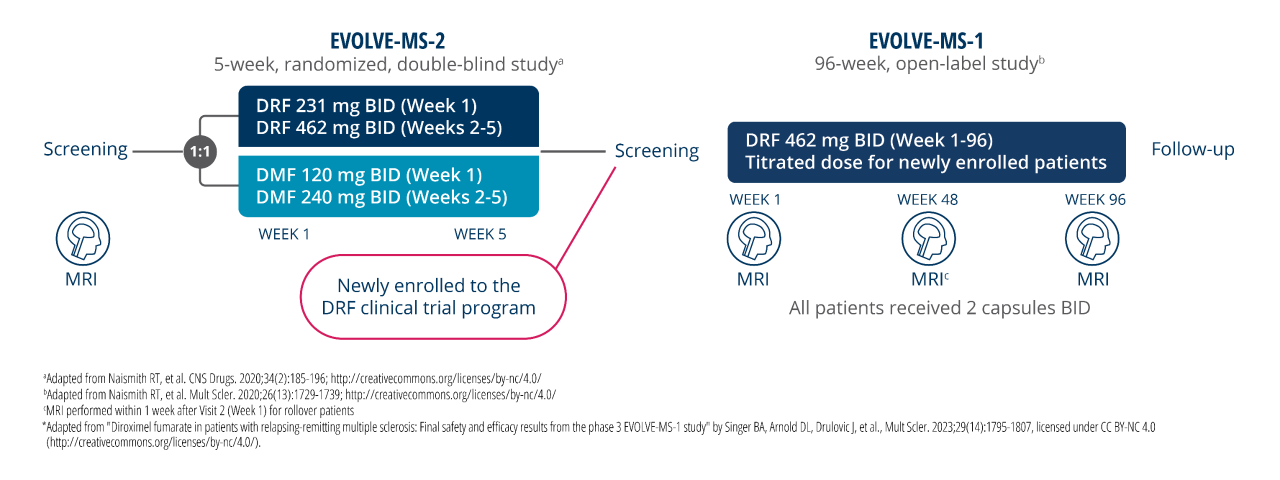
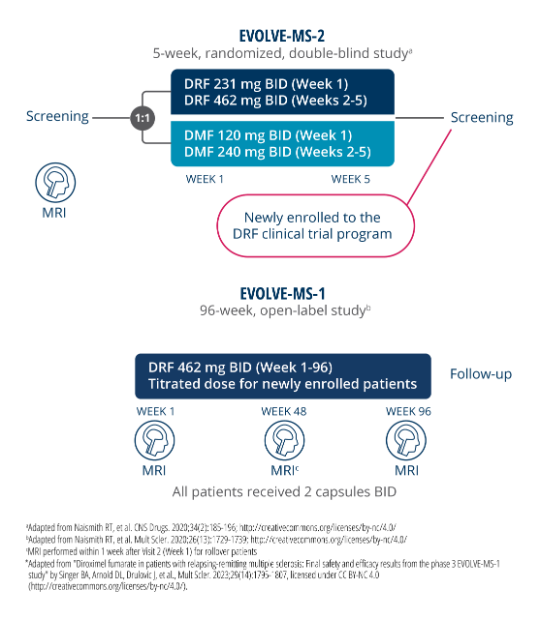
NEDA-3 was defined as having no relapse, no 24-week confirmed disability progression (CDP), and no MRI activity (ie, no new or newly enlarging T2 lesions and no new gadolinium-enhancing lesions)1
Limitations1:
- Due to the study design, disease activity within the first 7 weeks before re-baselining could not be assessed in these patients
- This was a post hoc analysis and NEDA-3 was an exploratory efficacy endpoint of the EVOLVE-MS-1 study. The clinical and radiological efficacy endpoints that comprise NEDA-3 have been studied individually in DMF and VUMERITY, but not with NEDA-3 named as a specific primary or secondary trial endpoint
More than half of patients on VUMERITY achieved No Evidence of Disease Activity
(NEDA-3) after re-baselining1
NEDA-3 Outcomes After Re-baselining in VUMERITY Rollover Patients
Week 96 prior DRF group


- Corresponding estimates at week 96 were 48.2% in the prior DMF group, and 36.5% in the de novo group
- Compared with the de novo group, the proportion of patients achieving NEDA over the study period was higher in the prior DRF group and the prior DMF group
*NEDA-3 was defined as having no relapse, no 24-week CDP, no new or newly enlarging T2 lesions, and no new Gd+ lesions.
Patients across all groups experienced adverse events; most were mild to moderate1
Adverse events occurred in 212 (88.7%) patients in the prior DRF group, 207 (92.0%) patients in the prior DMF group, and 519 (87.5%) patients in the de novo group; most were mild to moderate in severity
- The most common AEs in the prior DRF and prior DMF groups were MS relapse (20.1% and 20.0%, respectively), upper respiratory tract infection (17.6% and 15.6%), lymphopenia (14.6% and 16.9%), and flushing (13.8% and 12.9%); in the de novo group, the most common AEs were flushing (38.1%), MS relapse (19.1%), and nasopharyngitis (14.5%)
- Lymphopenia was reported in 51 (8.6%) patients in the de novo group
- Adverse events led to discontinuation of DRF in 23 (9.6%) patients in the prior DRF group, 13 (5.8%) patients in the prior DMF group, and 49 (8.3%) patients in the de novo group
- Serious AEs occurred in 12.1% (n=29) of patients in the prior DRF group, 11.1% (n=25) of patients in the prior DMF group, and 11.6% (n=69) of patients in the de novo group
- There were 71 (29.7%) discontinuations in the prior DRF group, 61 (27.1%) in the prior DMF group, and 125 (21.1%) in the de novo group
Baseline Demographics and Disease Characteristics in DRF
Rollover Patients
Baseline characteristics were generally consistent between the patient groups.
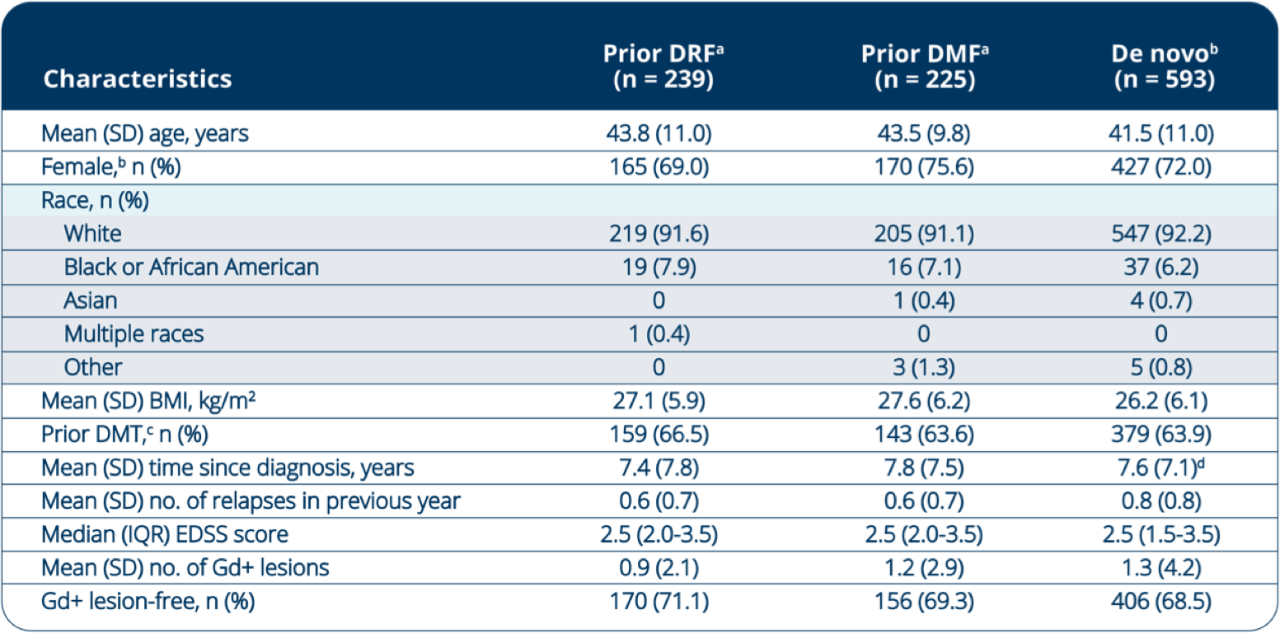
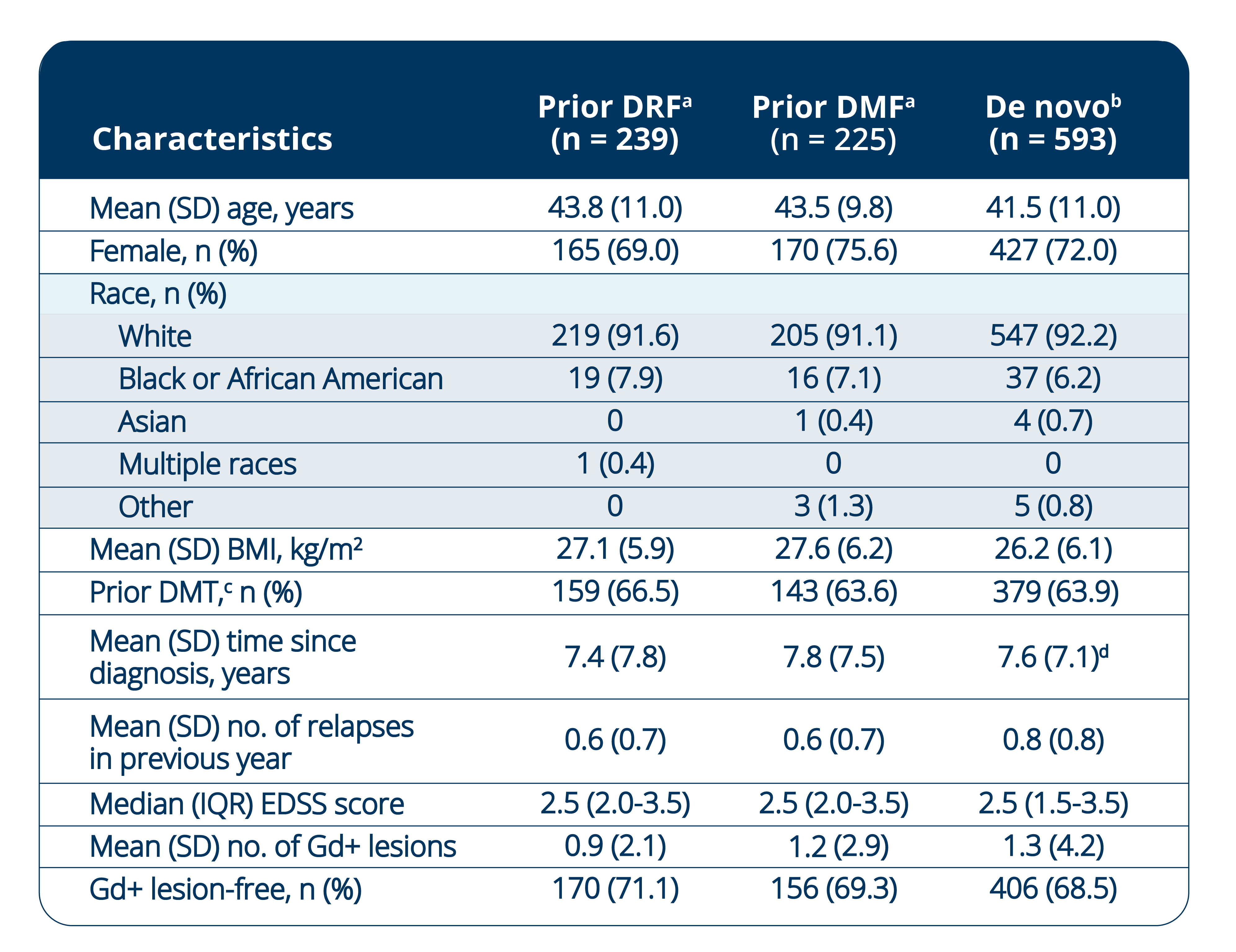
ᵃEVOLVE-MS-2 baseline measurements.
bBaseline demographics and disease characteristics for the de novo group were previously reported in: Singer BA, Arnold DL, Drulovic J, et al. Diroximel fumarate in patients with relapsing-remitting multiple sclerosis: final safety and efficacy results from the phase 3 EVOLVE-MS-1 study. Mult Scler. 2023;29(14):1795–1807. doi:10.1177/13524585231205708
cPrior DMT includes immunomodulatory and immunosuppressant (investigational or approved).
dn=592.